

In our study, we show how a Transformer language model, trained simply to predict a masked (hidden) amino acid in a protein sequence, recovers high-level structural and functional properties of proteins through its attention mechanism. We demonstrate that attention (1) captures the folding structure of proteins, connecting regions that are apart in the underlying sequence but spatially close in the protein structure, and (2) targets binding sites, a key functional component of proteins. We also introduce a three-dimensional visualization of the interaction between attention and protein structure. Our findings align with biological processes and provide a tool to aid scientific discovery. The code for the visualization tool and experiments is available at https://github.com/salesforce/provis.
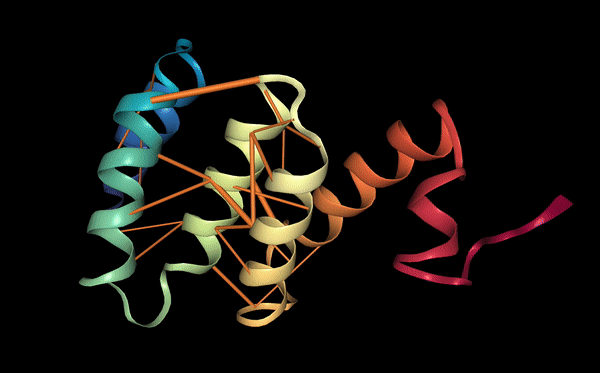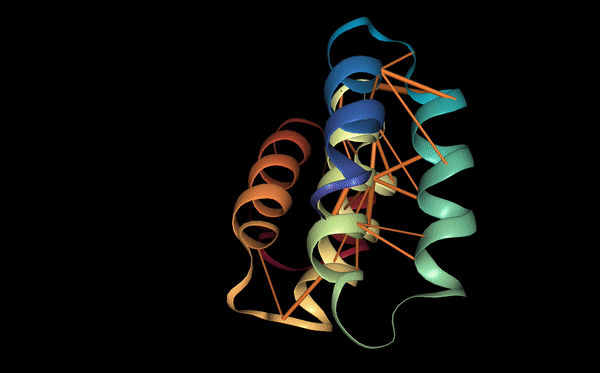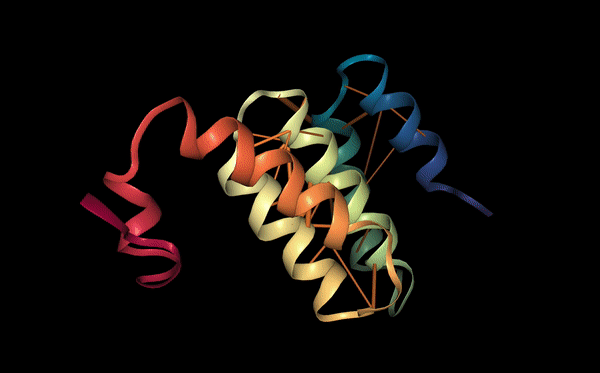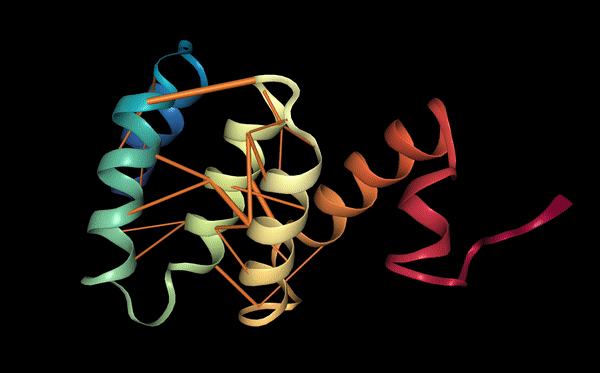
Protein Language Models
Proteins are complex molecules that play a critical functional and structural role for all forms of life on this planet. The study of proteins has led to many advances in disease therapies, and the application of machine learning to proteins has the potential for far-reaching applications in medicine and beyond.
Though the behavior of proteins is complex, the underlying representation is quite simple; every protein can be expressed as a chain of amino acids:

Abstracted as a sequence of discrete symbols, proteins may be modeled using machine learning architectures developed for natural language. In particular, the Transformer model [3], which has recently revolutionized the field of Natural Language Processing (NLP) shows promise for a similar impact on protein modeling. However, the Transformer presents challenges in interpretability due to the complexity of its underlying architecture. In the following sections, we show how to use attention as a kind of lens into the inner workings of this “black box” model.
Attention in the Transformer model
The defining characteristic of the Transformer is its attention mechanism, which we’ll illustrate with an example from natural language. Suppose we train a Transformer to predict the next word in a sentence — a task known as language modeling:

For the model to guess a reasonable next word, e.g. “rolled”, it must understand that “it” refers to “ball” (a relationship known as coreference). The Transformer does this by looking back at, or attending to, the earlier mention of “ball” when processing “it”:

Thus attention is a way for the model to contextualize a word by directly relating it to other words from the input. This contrasts with the traditional recurrent neural network — a predecessor of the Transformer — which abstracts all of the previous context words into a single state vector.
Words attend to each other to varying degrees based on their respective attention weights. These weights range between 0 and 1, where 0 indicates that a word has no direct relationship with the other word, while 1 indicates a strong dependency. In the above example, the attention weight might represent the model’s confidence that “it” refers to “ball”. The model may also attend to other words, capturing different types of relationships.
Note that the Transformer learns how to focus its attention. Before training, it has no concept of syntax or semantics — words are merely disconnected symbols. But when we train the model with a language modeling objective, we are forcing it to learn internal representations that enable it to understand the rules of natural language.
We can then transfer the model’s knowledge of language to more practical tasks, e.g., sentiment detection, by fine-tuning the models on these downstream tasks [4]. The advantage of first pre-training the model on a language modeling objective is that we can leverage the vast amount of available unlabeled text since the words themselves serve as the labels. This is known as self-supervised learning.
The language of proteins
By representing proteins as sequences of discrete symbols (amino acids), we can apply the same language modeling pre-training strategy to leverage the ~280 million available unlabeled protein sequences [5]. In this case, the model is trained to predict an amino acid instead of a word:

Studies have shown that language model pre-training improves the performance of the Transformer on protein classification [6] and generation [7] tasks. See this post on the ProGen model to learn more about the latter.
What does attention understand about proteins?
So what do protein models actually learn from pre-training? We attempt to answer this question by analyzing the attention patterns in the BERT [8] Transformer model from TAPE [6], which was pre-trained on language modeling of 31 million protein sequences [9]. BERT uses masked language modeling, where the model learns to predict masked (missing) tokens in the input:

The TAPE model uses the BERT-Base architecture, which has 12 layers, each of which has 12 distinct attention mechanisms, or heads. In the following sections, we explore how specific attention heads capture different structural and functional properties of proteins.
Protein structure: contact maps
Though a protein may be abstracted as a sequence of amino acids, it represents a physical entity with a well-defined three-dimensional structure. One way to characterize this structure is by a contact map, which describes the pairs of amino acids that are in contact (within 8 angstroms, or 8 x 10-10 m, of one another) in the folded protein structure, but lie apart (by at least 6 positions) in the underlying sequence:

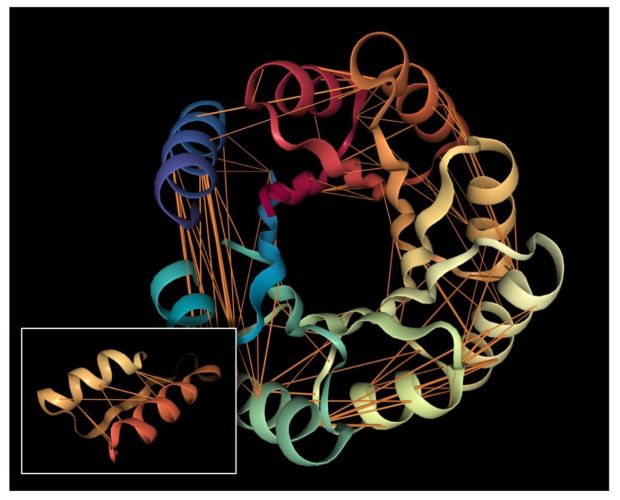
We explored the degree to which attention captures these contact relationships by analyzing the attention patterns of 5,000 protein sequences and comparing them to ground-truth contact maps. Our analysis revealed that one particular head — the 12th layer’s 4th head, denoted as head 12-4 — aligned remarkably well with the contact map. For “high confidence” attention (> .9 ), 76% of this head’s total attention connected amino acids that were in contact. In contrast, the background frequency of contacts among all amino acid pairs in the dataset is just 1.3%.
Below we visualize the attention from head 12-4, superimposed on the three-dimensional protein structure (though the model itself is provided with no spatial information), showing how the attention targets spatially close amino acids:

As a predictor of contact maps, Head 12-4 is well-calibrated: the attention weight approximates the actual probability of two amino acids being in contact, as shown in the figure below.

Considering the model was trained with a masked language modeling objective with no spatial information in its inputs or training labels, the presence of a head that identifies contacts is surprising. One potential reason for this localizing behavior could be that contacts are more likely to biochemically interact with one another, thereby constraining the amino acids that may occupy these positions. In a language model, therefore, knowing such contacts of masked tokens could provide valuable context for amino acid prediction. The acquired spatial understanding can, in turn, aid downstream tasks such as protein interaction prediction.
Protein function: binding sites
Proteins may also be characterized by their functional properties. Binding sites are regions of protein sequences that bind with other molecules to carry out a specific function. For example, the HIV-1 protease is an enzyme responsible for a critical process in the replication of HIV [10]. It has a binding site, shown in the figure below, which is a target for drug development to ensure inhibition.

Following a similar approach to our analysis of contact maps, we discovered several attention heads that specifically targeted binding sites. For example, head 7-1 (shown above) focused 44% of high confidence attention (>.9) on binding sites, which occur at only 4.8% of amino acid positions in the dataset. We also found that binding sites are often targeted from far away in the sequence. In head 7-1, just discussed, the average distance spanned by attention to binding sites is 124 positions.
Why does attention target binding sites? Proteins largely function to bind to other molecules, whether small molecules, proteins, or other macromolecules. Past work has shown that binding sites can reveal evolutionary relationships among proteins [11] and that particular structural motifs in binding sites are mainly restricted to specific families or superfamilies of proteins [12]. Thus binding sites provide a high-level characterization of the protein that may be relevant for the model to understand the sequence as a whole. An understanding of binding sites can, in turn, help the model with downstream tasks related to protein function, e.g., enzyme optimization.
Looking forward
In contrast to NLP, which seeks to automate a capability that humans already possess — understanding natural language — protein modeling seeks to shed light on biological processes that are not yet fully understood. By analyzing the differences between the model’s representations and our current understanding of proteins, we may be able to gain insights that fuel scientific discovery. But in order for learned representations to be accessible to domain experts, they must be presented in an appropriate context, e.g., embedded within protein structure as shown above. We believe there is a great potential to develop a wide variety of such contextual visualizations of learned representations in biology and other scientific domains.
We will be presenting this work at the ICML Workshop on ML Interpretability for Scientific Discovery on July 17th.
Resources
Paper: BERTology Meets Biology: Interpreting Attention in Protein Language Models
Code: https://github.com/salesforce/provis
When referencing this work, please cite:
@misc{vig2020bertology,
title={BERTology Meets Biology: Interpreting Attention in Protein Language Models},
author={Jesse Vig and Ali Madani and Lav R. Varshney and Caiming Xiong and Richard Socher and Nazneen Fatema Rajani},
year={2020},
eprint={2006.15222},
archivePrefix={arXiv},
primaryClass={cs.CL},
url={https://arxiv.org/abs/2006.15222}
}Acknowledgements
This work was done in collaboration with Ali Madani, Lav Varshney, Caiming Xiong, and Richard Socher. Thanks to Tong Niu, Stephan Zheng, Victoria Lin, and Melvin Gruesbeck for their valuable feedback .
References
[1] Rose, Alexander, et al. “NGL Viewer: a web application for molecular visualization.” Nucleic Acids Research, 43(W1):W576–W579. 2015.
[2] Rose, Alexander, et al. “NGL viewer: web-based molecular graphics for large complexes.” Bioinformatics, 34(21):3755–3758. 2018.
[3] Vaswani, Ashish, et al. “Attention is All You Need.” In NeurIPS 2017.
[4] Howard, Jeremy, et al. “Universal Language Model Fine-Tuning for Text Classification.” In ACL 2018.
[5] Alquraishi, Mohammed. “The Future of Protein Science will not be Supervised”. https://moalquraishi.wordpress.com/2019/04/01/the-future-of-protein-science-will-not-be-supervised/ (2019).
[6] Rao, Roshan, et al. “Evaluating protein transfer learning with TAPE.” In NeurIPS 2019.
[7] Madani, Ali, et al. “ProGen: Language modeling for protein generation.” bioRxiv. 2020.
[8] Devlin, Jacob, et al. “BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding.” In NAACL 2019.
[9] El-Gebali, Sara, et al. “The Pfam protein families database in 2019.” Nucleic Acids Research, 47(D1):D427–D432. 2019.
[10] Brik, Ashraf, et al. “HIV-1 protease: mechanism and drug discovery.” Organic and Biomolecular Chemistry 1(1):5–14. 2003.
[11] Lee, Juyong, et al. “Global organization of a binding site network gives insight into evolution and structure-function relationships of proteins.” Sci Rep, 7(11652). 2017.
[12] Kinjo, Akira, et al. “Comprehensive structural classification of ligand-binding motifs in proteins.” Structure, 17(2). 2009.























